
The excess electron at polyethylene interfaces†
 a
a
Abstract
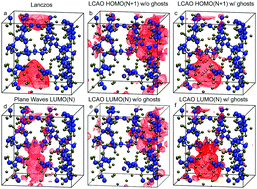
This work investigates the energy and spatial properties of excess electrons in polyethylene in bulk phases and, for the first time, at amorphous vacuum interfaces using a pseudopotential single-electron method (Lanczos diagonalisation) and density functional theory (DFT). DFT calculations are made employing two approaches: with pseudopotentials/plane waves and the local-density approximation; and with all-electron Gaussian basis functions at the B3LYP level of theory, supplemented with a lattice of ghost atoms. All three methods predict similar spatial localisation of the excess electron, but a reliable comparison of its energy can only be made between the Lanczos and DFT using Gaussian bases. While Lanczos predicts that an excess electron would preferentially localise in nanovoids with diameters smaller than 1 nm, DFT suggests that it would localise on surfaces in nanovoids larger than 1 nm. Overall we conclude that in DFT studies of polyethylene/vacuum interfaces at the current level of theory, orbital-based methods provide a useful representation of excess electron properties.
 Please wait while we load your content...
Please wait while we load your content...

