
Combined quantum mechanical and molecular mechanical method for metal–organic frameworks: proton topologies of NU-1000†

Abstract
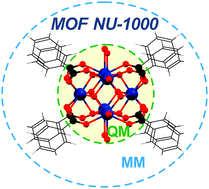
Metal–organic frameworks (MOFs) are materials with applications in catalysis, gas separations, and storage. Quantum mechanical (QM) calculations can provide valuable guidance to understand and predict their properties. In order to make the calculations faster, rather than modeling these materials as periodic (infinite) systems, it is useful to construct finite models (called cluster models) and use subsystem methods such as fragment methods or combined quantum mechanical and molecular mechanical (QM/MM) methods. Here we employ a QM/MM methodology to study one particular MOF that has been of widespread interest because of its wide pores and good solvent and thermal stability, namely NU-1000, which contains hexanuclear zirconium nodes and 1,3,6,8-tetrakis(p-benzoic acid)pyrene (TBAPy4−) linkers. A modified version of the Bristow–Tiana–Walsh transferable force field has been developed to allow QM/MM calculations on NU-1000; we call the new parametrization the NU1T force field. We consider isomeric structures corresponding to various proton topologies of the [Zr6(μ3-O)8O8H16]8+ node of NU-1000, and we compute their relative energies using a QM/MM scheme designed for the present kind of problem. We compared the results to full quantum mechanical (QM) energy calculations and found that the QM/MM models can reproduce the full QM relative energetics (which span a range of 334 kJ mol−1) with a mean unsigned deviation (MUD) of only 2 kJ mol−1. Furthermore, we found that the structures optimized by QM/MM are nearly identical to their full QM optimized counterparts.
 Please wait while we load your content...
Please wait while we load your content...

