
The structures of liquid pyridine and naphthalene: the effects of heteroatoms and core size on aromatic interactions†
 *a
N. T.
Skipper
*b
*a
N. T.
Skipper
*b
Abstract
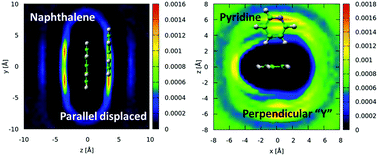
Total neutron scattering has been used in conjunction with H/D and *N/15N isotopic substitution to determine the detailed liquid-state structures of pyridine and naphthalene. Analysis of the data via an empirical potential-based structure refinement method has allowed us to interrogate the full six-dimensional spatial and orientational correlation surfaces in these systems, and thereby to deduce the fundamental effects of a heteroatom and aromatic core-size on intermolecular π–π interactions. We find that the presence of a nitrogen heteroatom, and concomitant dipole moment, in pyridine induces surprisingly subtle departures from the structural correlations observed in liquid benzene: in both cases the most probable local motif is based on perpendicular (edge-to-face) intermolecular contacts, while parallel-displaced configurations give rise to a clear shoulder in the correlation surface. However, the effect of the heteroatom is revealed through detailed analysis of the intermolecular orientational correlations. This analysis shows a tendency for neighbouring pyridine molecules to direct one meta- and one para-hydrogen towards the neighbouring aromatic π-orbitals in edge-to-face configurations, while head-to-tail alignment of adjacent nitrogen atoms is favoured in face-to-face configurations. In contrast to this, increasing aromatic core size from one to only two rings has a clear and profound effect on the π–π interactions and liquid structure. Our experiments show that naphthalene–naphthalene contacts are dominated by parallel-displaced configurations, akin to those found in graphite. This marks a fundamental difference with the structure of liquid benzene, in which perpendicular geometries are favoured. Furthermore, it is remarkable to note that in the systems studied, the most favoured spatio-orientational configurations observed in the liquid state are not predicted from ab initio calculations and/or solid state crystallographic studies. This highlights the need for caution when extrapolating the results of crystallographic and computational studies to aromatic interactions in liquids and disordered systems.
 Please wait while we load your content...
Please wait while we load your content...

