
Tailoring electronic properties of multilayer phosphorene by siliconization†

Abstract
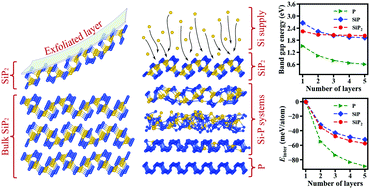
Controlling the thickness dependence of electronic properties for two-dimensional (2d) materials is among the primary goals for their large-scale applications. Herein, employing a first-principles computational approach, we predict that Si interaction with multilayer phosphorene (2d-P) can result in the formation of highly stable 2d-SiP and 2d-SiP2 compounds with a weak interlayer interaction. Our analysis demonstrates that these systems are semiconductors with band gap energies that can be governed by varying the thicknesses and stacking arrangements. Specifically, the siliconization of phosphorene allows designing 2d-SiPx materials with a significantly weaker thickness dependence of electronic properties than that in 2d-P and to develop ways for their tailoring. We also reveal the spatial dependence of electronic properties for 2d-SiPx highlighting the difference in the effective band gaps for different layers. Particularly, our results show that the central layers in the multilayer 2d systems determine their overall electronic properties, while the role of the outermost layers is noticeably smaller.
 Please wait while we load your content...
Please wait while we load your content...

