
NEXAFS spectroscopy of ionic liquids: experiments versus calculations†
 a
Richard P.
Matthews,
a
Matthew T.
Clough,
a
Claire R.
Ashworth,
a
Agnieszka
Brandt-Talbot,
b
Paul J.
Corbett,
b
Robert G.
Palgrave,
c
Richard A.
Bourne,
de
Thomas W.
Chamberlain,
e
Tom
Vander Hoogerstraete,
f
Paul B. J.
Thompson,
gh
Patricia A.
Hunt,
a
Nicholas A.
Besley
*i
and
Kevin R. J.
Lovelock
*j
a
Richard P.
Matthews,
a
Matthew T.
Clough,
a
Claire R.
Ashworth,
a
Agnieszka
Brandt-Talbot,
b
Paul J.
Corbett,
b
Robert G.
Palgrave,
c
Richard A.
Bourne,
de
Thomas W.
Chamberlain,
e
Tom
Vander Hoogerstraete,
f
Paul B. J.
Thompson,
gh
Patricia A.
Hunt,
a
Nicholas A.
Besley
*i
and
Kevin R. J.
Lovelock
*j
Abstract
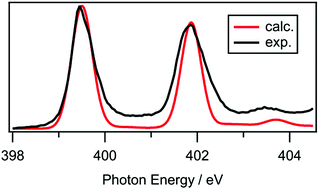
Experimental near edge X-ray absorption fine structure (NEXAFS) spectra are reported for 12 ionic liquids (ILs) encompassing a range of chemical structures for both the sulfur 1s and nitrogen 1s edges and compared with time-dependent density functional theory (TD-DFT) calculations. The energy scales for the experimental data were carefully calibrated against literature data. Gas phase calculations were performed on lone ions, ion pairs and ion pair dimers, with a wide range of ion pair conformers considered. For the first time, it is demonstrated that TD-DFT is a suitable method for simulating NEXAFS spectra of ILs, although the number of ions included in the calculations and their conformations are important considerations. For most of the ILs studied, calculations on lone ions in the gas phase were sufficient to successfully reproduce the experimental NEXAFS spectra. However, for certain ILs – for example, those containing a protic ammonium cation – calculations on ion pairs were required to obtain a good agreement with experimental spectra. Furthermore, significant conformational dependence was observed for the protic ammonium ILs, providing insight into the predominant liquid phase cation–anion interactions. Among the 12 investigated ILs, we find that four have an excited state that is delocalised across both the cation and the anion, which has implications for any process that depends on the excited state, for example, radiolysis. Considering the collective experimental and theoretical data, we recommend that ion pairs should be the minimum number of ions used for the calculation of NEXAFS spectra of ILs.
 Please wait while we load your content...
Please wait while we load your content...

