
The hydration structure of the heavy-alkalines Rb+ and Cs+ through molecular dynamics and X-ray absorption spectroscopy: surface clusters and eccentricity†

Abstract
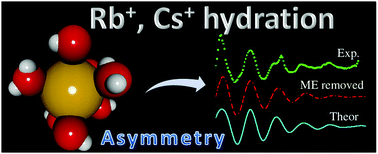
Physicochemical properties of the two heaviest stable alkaline cations, Rb+ and Cs+, in water have been examined from classical molecular dynamics (MD) simulations. Alkaline cation–water intermolecular potentials have been built from ab initio interaction energies of [M(H2O)n]+ clusters. Unlike in the case of other monatomic metal cations, the sampling needed the inclusion of surface clusters to properly describe the interactions. The first coordination shell is found at an average M–O distance of 2.87 Å and 3.12 Å for Rb+ and Cs+, respectively, with coordination numbers of 8 and 10. Structural, dynamical and energetic properties are discussed on the basis of the delicate compromise among the ion–water and water–water interactions which contribute almost on the same foot to the definition of the solvent structure around the ions. A significant asymmetry is detected in the Rb+ and Cs+ first hydration shell. Reorientational times of first-shell water molecules for Cs+ support a clear structure-breaking nature for this cation, whereas the Rb+ values do not differ from pure water behavior. Experimental EXAFS and XANES spectra have been compared to simulated ones, obtained by means of application of the FEFF code to a set of statistically significant structures taken from the MD simulations. Due to the presence of multi-excitations in the absorption spectra, theoretical–experimental agreement for the EXAFS spectra is reached when the multi-excitations are removed from the experimental spectra.
 Please wait while we load your content...
Please wait while we load your content...

