
Stacking interactions involving non-Watson–Crick basepairs: dispersion corrected density functional theory studies†
 ab
and
Dhananjay
Bhattacharyya
*ab
ab
and
Dhananjay
Bhattacharyya
*ab
Abstract
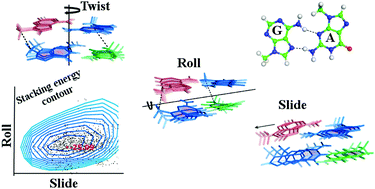
The G:A basepair, stabilized by hydrogen bonding through the sugar edge of guanine and Hoogsteen edge of adenine in trans orientation (G:A S:HT), appears very frequently in the solved RNA structures and is very stable. We have carried out stacking energy analyses of two sequences, namely C:G W:WC::G:A S:HT and G:C W:WC::G:A S:HT (‘:’ represents basepairing and ‘::’ represents stacking interactions), formed by this non-Watson–Crick basepair, by DFT-D. We have scanned nearly the complete phase space by modeling structures of these sequences using different values of the basepair orientation parameters to determine the most stable conformations. It is found that the most stable conformations are rather far from the most frequently found orientations. We have considered the effect of sugar–phosphate backbone connectivity as an energy penalty arising from deformation of pseudo bond lengths between C1′ atoms of successive bases along the strands. Augmentation of stacking energy from DFT-D by this coarse grain energy gives predicted structures extremely similar to the experimentally determined ones. It has been observed that the best stacking with small twist values is associated with positive roll and negative slide values, which are similar to their values in A-RNA structures for most sequences. Among the two base pair steps, C:G W:WC::G:A S:HT appears to be more stable in terms of stacking energy as compared to G:C W:WC::G:A S:HT possibly due to larger stacking overlap in the former one.
 Please wait while we load your content...
Please wait while we load your content...

