
Theoretical investigation of the solid–liquid phase transition in protonated water clusters†

Abstract
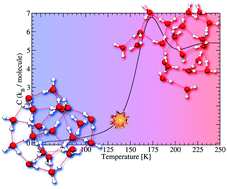
Protonated water clusters have received a lot of attention as they offer tools to bridge the gap between molecular and bulk scales of water. However, their properties are still not fully understood and deserve further theoretical and experimental investigations. In this work, we simulate the caloric curves of protonated water clusters (H2O)nH+ (n = 20–23). These curves, which have recently been measured experimentally, are characteristic of the phase changes occurring in the aggregates with respect to temperature. The present simulations are achieved by combining parallel-tempering molecular dynamics and the self-consistent-charge density-functional based tight-binding approach and are focused on a restricted size range around (H2O)21H+ which presents singular properties. The shape of the experimental caloric curves and their size dependence are satisfactorily reproduced by the simulations which allows us to further provide a description of the phase transition in terms of structural modifications, dynamics of water molecules and proton mobility. Similar to the experiments, we observe that (H2O)21H+ exhibits a sharper phase transition than the neighbouring size clusters, which can be traced back to both structural and dynamic peculiarities.
 Please wait while we load your content...
Please wait while we load your content...

