
Kinetics and dynamics of the C(3P) + H2O reaction on a full-dimensional accurate triplet state potential energy surface

Abstract
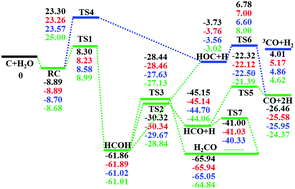
A full dimensional accurate potential energy surface (PES) for the C(3P) and H2O reaction is developed based on ∼34 000 data points calculated at the level of the explicitly correlated unrestricted coupled cluster method with single, double, and perturbative triple excitations with the augmented correlation-consistent polarized triple zeta basis set (CCSD(T)-F12a/AVTZ). The PES is invariant with respect to the permutation of the two hydrogen atoms and the total root mean square error (RMSE) of the fit is only 0.31 kcal mol−1. The PES features two barriers in the entrance channel and several potential minima, as well as multiple product channels. The rate coefficients of this reaction calculated using a transition-state theory and quasi-classical trajectory (QCT) method are small near room temperature, consistent with experiments. The reaction dynamics is also investigated with QCT on the new PES, which found that the reactivity is constrained by the entrance barriers and the final product branching is not statistical.
 Please wait while we load your content...
Please wait while we load your content...

