
The borazine dimer: the case of a dihydrogen bond competing with a classical hydrogen bond
 *a
and
*a
and
Abstract
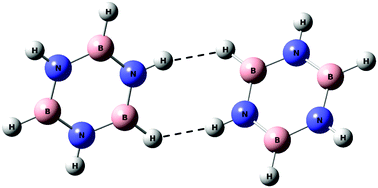
Dimers of borazine were studied using matrix isolation infrared spectroscopy and ab initio quantum chemical calculations. Computations were performed at the MP2 and M06-2X levels of theory using the 6-311++G(d,p) and aug-cc-pVDZ basis sets for the various homodimers. At both levels of theory, an aligned stacked structure was found to be the global minimum, which was nearly isoenergetic to a parallel displaced structure. A T-shaped structure, where the N–H of one borazine pointed towards the N of the second borazine, was found to be a local minimum. In addition to these structures, a dihydrogen bonded structure, where the hydrogen attached to the nitrogen of borazine interacted with the hydrogen attached to the boron atom of another borazine, was also indicated. Experimentally, we observed the T-shaped dimer and the dihydrogen bonded dimer. This is one of the rare examples of experimental evidence for a dihydrogen bond, in a system other than in a metal hydride. These results for the borazine dimer were clearly different from the benzene dimer where the parallel displaced structure was found to be the global minimum followed by the T-shaped structure at the MP2/aug-cc-pVDZ level of theory. AIM, EDA and NBO analyses were carried out for all the structures to explore the nature of interactions.
 Please wait while we load your content...
Please wait while we load your content...

