
A non-empirical calculation of 2p core-electron excitation in compounds with 3d transition metal ions using ligand-field and density functional theory (LFDFT)†
 *a
and
*a
and
Abstract
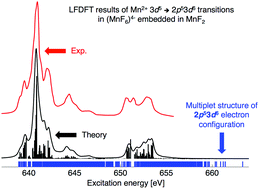
Methodological advents for the calculation of the multiplet energy levels arising from multiple-open-shell 2p53dn+1 electron configurations, with n = 0, 1, 2,… and 9, are presented. We use the Ligand-Field Density Functional Theory (LFDFT) program, which has been recently implemented in the Amsterdam Density Functional (ADF) program package. The methodology consists of calculating the electronic structure of a central metal ion together with its ligand coordination by means of the Density Functional Theory code. Besides, the core-hole effects are treated by incorporating many body effects and corrections via the configuration interaction algorithm within the active space of Kohn–Sham orbitals with dominant 2p and 3d characters of the transition metal ions, using an effective ligand-field Hamiltonian. The Slater–Condon integrals (F2(3d,3d), F4(3d,3d), G1(2p,3d), G3(2p,3d) and F2(2p,3d)), spin–orbit coupling constants (ζ2p and ζ3d) and parameters of the ligand-field potential (represented within the Wybourne formalism) are therefore determined giving rise to the multiplet structures of systems with 3dn and 2p53dn+1 configurations. The oscillator strengths of the electric-dipole allowed 3dn → 2p53dn+1 transitions are also calculated allowing the theoretical simulation of the absorption spectra of the 2p core-electron excitation. This methodology is applied to transition metal ions in the series Sc2+, Ti2+,…, Ni2+ and Cu2+ but also to selective compounds, namely SrTiO3 and MnF2. The comparison with available experimental data is good. Therefore, a non-empirical ligand-field treatment of the 2p53dn+1 configurations is established and available in the ADF program package illustrating the spectroscopic details of the 2p core-electron excitation that can be valuable in the further understanding and interpretation of the transition metal L2,3-edge X-ray absorption spectra.
 Please wait while we load your content...
Please wait while we load your content...

