
Pair natural orbital and canonical coupled cluster reaction enthalpies involving light to heavy alkali and alkaline earth metals: the importance of sub-valence correlation†

Abstract
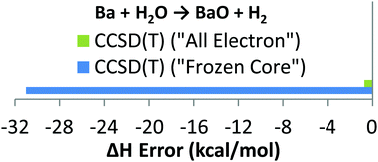
In this work, we tested canonical and domain based pair natural orbital coupled cluster methods (CCSD(T) and DLPNO-CCSD(T), respectively) for a set of 32 ligand exchange and association/dissociation reaction enthalpies involving ionic complexes of Li, Be, Na, Mg, Ca, Sr, Ba and Pb(II). Two strategies were investigated: in the former, only valence electrons were included in the correlation treatment, giving rise to the computationally very efficient FC (frozen core) approach; in the latter, all non-ECP electrons were included in the correlation treatment, giving rise to the AE (all electron) approach. Apart from reactions involving Li and Be, the FC approach resulted in non-homogeneous performance. The FC approach leads to very small errors (<2 kcal mol−1) for some reactions of Na, Mg, Ca, Sr, Ba and Pb, while for a few reactions of Ca and Ba deviations up to 40 kcal mol−1 have been obtained. Large errors are both due to artificial mixing of the core (sub-valence) orbitals of metals and the valence orbitals of oxygen and halogens in the molecular orbitals treated as core, and due to neglecting core–core and core–valence correlation effects. These large errors are reduced to a few kcal mol−1 if the AE approach is used or the sub-valence orbitals of metals are included in the correlation treatment. On the technical side, the CCSD(T) and DLPNO-CCSD(T) results differ by a fraction of kcal mol−1, indicating the latter method as the perfect choice when the CPU efficiency is essential. For completely black-box applications, as requested in catalysis or thermochemical calculations, we recommend the DLPNO-CCSD(T) method with all electrons that are not covered by effective core potentials included in the correlation treatment and correlation-consistent polarized core valence basis sets of cc-pwCVQZ(-PP) quality.
 Please wait while we load your content...
Please wait while we load your content...

