
Stabilization of carbocations CH3+, C2H5+, i-C3H7+, tert-Bu+, and cyclo-pentyl+ in solid phases: experimental data versus calculations†
 *ab
and
Anton S.
Nizovtsev
bc
*ab
and
Anton S.
Nizovtsev
bc
Abstract
Comparison of experimental infrared (IR) spectra of the simplest carbocations (with the weakest carborane counterions in terms of basicity, CHB11Hal11−, Hal = F, Cl) with their calculated IR spectra revealed that they are completely inconsistent, as previously reported for the t-Bu+ cation [Stoyanov E. S., et al. J. Phys. Chem. A, 2015, 119, 8619]. This means that the generally accepted explanation of hyperconjugative stabilization of the carbocations should be revised. According to the theory, one CH bond (denoted as  ) from each CH3/CH2 group transfers its σ-electron density to the empty 2pz orbital of the sp2 C atom, whereas the σ-electron density on the other CH bonds of the CH3/CH2 group slightly increases. From experimental IR spectra it follows that donation of the σ-electrons from the
) from each CH3/CH2 group transfers its σ-electron density to the empty 2pz orbital of the sp2 C atom, whereas the σ-electron density on the other CH bonds of the CH3/CH2 group slightly increases. From experimental IR spectra it follows that donation of the σ-electrons from the 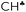 bond to the 2pz C-orbital is accompanied by equal withdrawal of the electron density from other CH bonds, that is, the electrons are supplied from each CH bond of the CH3/CH2 group. As a result, all CH stretches of the group are red shifted, and IR spectra show typical CH3/CH2 group vibrations. Experimental findings provided another clue to the electron distribution in the hydrocarbon cations and showed that the standard computational techniques do not allow researchers to explain a number of recently established features of the molecular state of hydrocarbon cations.
bond to the 2pz C-orbital is accompanied by equal withdrawal of the electron density from other CH bonds, that is, the electrons are supplied from each CH bond of the CH3/CH2 group. As a result, all CH stretches of the group are red shifted, and IR spectra show typical CH3/CH2 group vibrations. Experimental findings provided another clue to the electron distribution in the hydrocarbon cations and showed that the standard computational techniques do not allow researchers to explain a number of recently established features of the molecular state of hydrocarbon cations.
 Please wait while we load your content...
Please wait while we load your content...

