
Modelling temperature-dependent properties of polymorphic organic molecular crystals†

Abstract
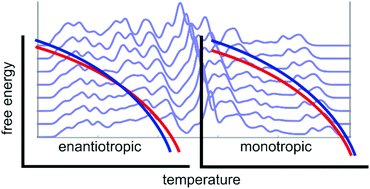
We present a large-scale study of the temperature-dependence of structures, free energy differences and properties of polymorphic molecular organic crystals. Lattice-vibrational Gibbs free energy differences between 475 pairs of polymorphs of organic molecular crystals have been calculated at 0 K and at their respective melting points, using a highly accurate anisotropic multipole-based force field and including thermal expansion through the use of a (negative) thermal pressure. Re-ranking of the relative thermodynamic stability of the polymorphs in each pair indicates the possibility of an enantiotropic phase transition between the crystal structures, which occurs in 21% of the studied systems. While vibrational contributions to free energies can have a significant effect on thermodynamic stability, the impact of thermal expansion on polymorph free energy differences is generally very small. We also calculate thermal expansion coefficients for the 864 crystal structures and investigate the temperature-dependence of mechanical properties, and pairwise differences in these properties between polymorphs.
 Please wait while we load your content...
Please wait while we load your content...

