
Density functional theory + U analysis of the electronic structure and defect chemistry of LSCF (La0.5Sr0.5Co0.25Fe0.75O3−δ)†
Abstract
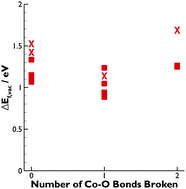
Reducing operating temperatures is a key step in making solid oxide fuel cell (SOFC) technology viable. A promising strategy for accomplishing this goal is employing mixed ion–electron conducting (MIEC) cathodes. La1−xSrxCo1−yFeyO3−δ (LSCF) is the most widely employed MIEC cathode material; however, rational optimization of the composition of LSCF requires fundamental insight linking its electronic structure to its defect chemistry. To provide the necessary insight, density functional theory plus U (DFT+U) calculations are used to investigate the electronic structure of LSCF (xSr = 0.50, yCo = 0.25). The DFT+U calculations show that LSCF has a significantly different electronic structure than La1−xSrxFeO3 because of the addition of cobalt, but that minimal electronic structure differences exist between La0.5Sr0.5Co0.25Fe0.75O3 and La0.5Sr0.5Co0.5Fe0.5O3. The oxygen vacancy 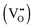 formation energy (ΔEf,vac) is calculated for
formation energy (ΔEf,vac) is calculated for 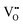 residing in different local environments within La0.5Sr0.5Co0.25Fe0.75O3. These results show that
residing in different local environments within La0.5Sr0.5Co0.25Fe0.75O3. These results show that 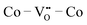 configurations have the highest ΔEf,vac, while
configurations have the highest ΔEf,vac, while 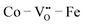 have the lowest ΔEf,vac and may act as traps for
have the lowest ΔEf,vac and may act as traps for 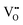 . We conclude that compositions with more Fe than Co are preferred because the additional
. We conclude that compositions with more Fe than Co are preferred because the additional 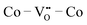 sites would lead to higher overall ΔEf,vac (and lower
sites would lead to higher overall ΔEf,vac (and lower 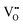 concentrations), while the trapping strength of the
concentrations), while the trapping strength of the 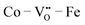 sites is relatively weak (∼0.3 eV).
sites is relatively weak (∼0.3 eV).
 Please wait while we load your content...
Please wait while we load your content...

