
Hunting for hydrogen: random structure searching and prediction of NMR parameters of hydrous wadsleyite†
 a
a
Abstract
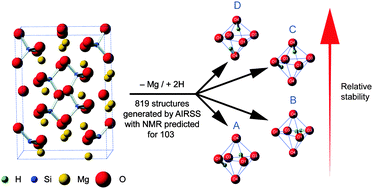
The structural chemistry of materials containing low levels of nonstoichiometric hydrogen is difficult to determine, and producing structural models is challenging where hydrogen has no fixed crystallographic site. Here we demonstrate a computational approach employing ab initio random structure searching (AIRSS) to generate a series of candidate structures for hydrous wadsleyite (β-Mg2SiO4 with 1.6 wt% H2O), a high-pressure mineral proposed as a repository for water in the Earth's transition zone. Aligning with previous experimental work, we solely consider models with Mg3 (over Mg1, Mg2 or Si) vacancies. We adapt the AIRSS method by starting with anhydrous wadsleyite, removing a single Mg2+ and randomly placing two H+ in a unit cell model, generating 819 candidate structures. 103 geometries were then subjected to more accurate optimisation under periodic DFT. Using this approach, we find the most favourable hydration mechanism involves protonation of two O1 sites around the Mg3 vacancy. The formation of silanol groups on O3 or O4 sites (with loss of stable O1–H hydroxyls) coincides with an increase in total enthalpy. Importantly, the approach we employ allows observables such as NMR parameters to be computed for each structure. We consider hydrous wadsleyite (∼1.6 wt%) to be dominated by protonated O1 sites, with O3/O4–H silanol groups present as defects, a model that maps well onto experimental studies at higher levels of hydration (J. M. Griffin et al., Chem. Sci., 2013, 4, 1523). The AIRSS approach adopted herein provides the crucial link between atomic-scale structure and experimental studies.
 Please wait while we load your content...
Please wait while we load your content...

