
Quantum and quasi-classical calculations for the S+ + H2(v,j) → SH+(v′,j′) + H reactive collisions
Abstract
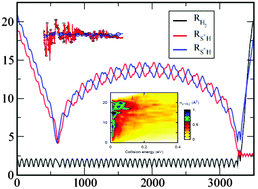
State-to-state cross-sections for the S+ + H2(v,j) → SH+(v′,j′) + H endothermic reaction are obtained using quantum wave packet (WP) and quasi-classical (QCT) methods for different initial ro-vibrational H2(v,j) over a wide range of translation energies. The final state distribution as a function of the initial quantum number is obtained and discussed. Additionally, the effect of the internal excitation of H2 on the reactivity is carefully studied. It appears that energy transfer among modes is very inefficient that vibrational energy is the most favorable for the reaction, and rotational excitation significantly enhances the reactivity when vibrational energy is sufficient to reach the product. Special attention is also paid to an unusual discrepancy between classical and quantum dynamics for low rotational levels while agreement improves with rotational excitation of H2. An interesting resonant behaviour found in WP calculations is also discussed and associated with the existence of roaming classical trajectories that enhance the reactivity of the title reaction. Finally, a comparison with the experimental results of Stowe et al. for S+ + HD and S+ + D2 reactions exhibits a reasonably good agreement with those results.
 Please wait while we load your content...
Please wait while we load your content...

