
How amino and nitro substituents direct electrophilic aromatic substitution in benzene: an explanation with Kohn–Sham molecular orbital theory and Voronoi deformation density analysis†
Abstract
The substituent effect of the amino and nitro groups on the electronic system of benzene has been investigated quantum chemically using quantitative Kohn–Sham molecular orbital theory and a corresponding energy decomposition analysis (EDA). The directionality of electrophilic substitution in aniline can accurately be explained with the amount of contribution of the 2pz orbitals on the unsubstituted carbon atoms to the highest occupied π orbital. For nitrobenzene, the molecular π orbitals cannot explain the regioselectivity of electrophilic substitution as there are two almost degenerate π orbitals with nearly the same 2pz contributions on the unsubstituted carbon atoms. The Voronoi deformation density analysis has been applied to aniline and nitrobenzene to obtain an insight into the charge rearrangements due to the substituent. This analysis method identified the orbitals involved in the C–N bond formation of the π system as the cause for the π charge accumulation at the ortho and para positions in the case of the NH2 group and the largest charge depletion at these same positions for the NO2 substituent. Furthermore, we showed that it is the repulsive interaction between the πHOMO of the phenyl radical and the πHOMO of the NH2 radical that is responsible for pushing up the πHOMO of aniline and therefore activating this π orbital of the phenyl ring towards electrophilic substitution.
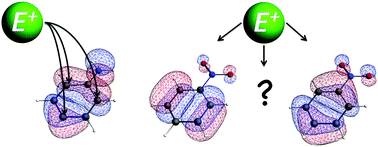
- This article is part of the themed collections: 2016 most accessed PCCP articles and Electron delocalization and aromaticity: 150 years of the Kekulé benzene structure
 Please wait while we load your content...
Please wait while we load your content...

