
Structural determination of niobium-doped silicon clusters by far-infrared spectroscopy and theory†
Abstract
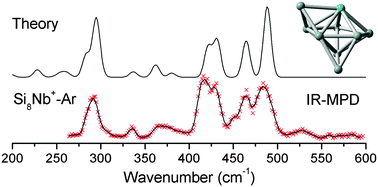
In this work, the structures of cationic SinNb+ (n = 4–12) clusters are determined using the combination of infrared multiple photon dissociation (IR-MPD) and density functional theory (DFT) calculations. The experimental IR-MPD spectra of the argon complexes of SinNb+ are assigned by comparison to the calculated IR spectra of low-energy structures of SinNb+ that are identified using the stochastic ‘random kick’ algorithm in conjunction with the BP86 GGA functional. It is found that the Nb dopant tends to bind in an apex position of the Sin framework for n = 4–9 and in surface positions with high coordination numbers for n = 10–12. For the larger doped clusters, it is suggested that multiple isomers coexist and contribute to the experimental spectra. The structural evolution of SinNb+ clusters is similar to V-doped silicon clusters (J. Am. Chem. Soc., 2010, 132, 15589–15602), except for the largest size investigated (n = 12), since V takes an endohedral position in Si12V+. The interaction with a Nb atom, with its partially unfilled 4d orbitals leads to a significant stability enhancement of the Sin framework as reflected, e.g. by high binding energies and large HOMO–LUMO gaps.
 Please wait while we load your content...
Please wait while we load your content...

