
Incorrect DFT-GGA predictions of the stability of non-stoichiometric/polar dielectric surfaces: the case of Cu2O(111)
Abstract
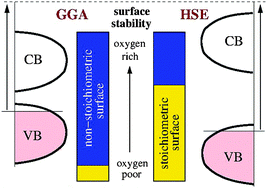
Scanning tunneling microscopy (STM) and hybrid density functional theory (DFT) have been used to study the stability and electronic characteristics of the Cu2O(111) surface. We challenge previous interpretations of its structure and composition and show that only appropriate (hybrid) calculations can correctly account for the relative thermodynamic stability of stoichiometric versus Cu-deficient terminations. Our theoretical finding of the stoichiometric surface to be most stable at oxygen-lean conditions is confirmed by an excellent matching between STM spectroscopy data and the calculated surface electronic structure. Beyond the specific case of the Cu2O(111) surface, and beyond the known deficiencies of GGA-based approaches in the description of oxide electronic structures, our work highlights the risk of an erroneous evaluation of the surface stability, in cases where the energetics and electronic characteristics are strongly coupled, as in a wide class of polar and/or non-stoichiometric oxide surfaces.
 Please wait while we load your content...
Please wait while we load your content...

