
Role of ligand–ligand vs. core–core interactions in gold nanoclusters†
Abstract
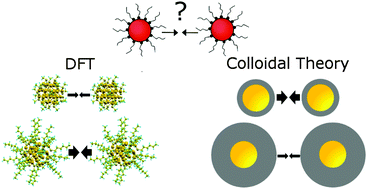
The controlled assembly of ligand-coated gold nanoclusters (NCs) into larger structures paves the way for new applications ranging from electronics to nanomedicine. Here, we demonstrate through rigorous density functional theory (DFT) calculations employing novel functionals accounting for van der Waals forces that the ligand–ligand interactions determine whether stable assemblies can be formed. The study of NCs with different core sizes, symmetry forms, ligand lengths, mutual crystal orientations, and in the presence of a solvent suggests that core-to-core van der Waals interactions play a lesser role in the assembly. The dominant interactions originate from combination of steric effects, augmented by ligand bundling on NC facets, and related to them changes in electronic properties induced by neighbouring NCs. We also show that, in contrast to standard colloidal theory approach, DFT correctly reproduces the surprising experimental trends in the strength of the inter-particle interaction observed when varying the length of the ligands. The results underpin the importance of understanding NC interactions in designing gold NCs for a specific function.
 Please wait while we load your content...
Please wait while we load your content...

