
Protonated thiophene-based oligomers as formed within zeolites: understanding their electron delocalization and aromaticity†
Abstract
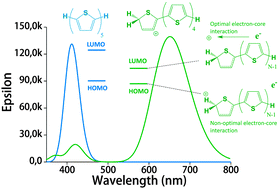
In an earlier work, protonated thiophene-based oligomers were identified inside ZSM-5 zeolites. The novel compounds exhibited π–π* absorption wavelengths deep within the visible region, earmarking them for possible use as chromophores in a variety of applications. In this computational study, we determine the factors that cause such low-energy transitions, and describe the electronic structure of these remarkable compounds. DFT calculations of conjugated thiophene-based oligomers with up to five monomer units reveal that the main absorption band of each protonated oligomer is strongly red-shifted compared to the unprotonated form. This effect is counterintuitive, since protonation is expected to diminish aromaticity, and thereby increase the HOMO–LUMO gap. We find that upon protonation the π-electrons remain delocalized over the entire π-conjugated molecule, but the positive charge is localized predominantly on the protonated side of the molecule. A possible explanation for this ground-state charge localization is the participation of the C–H bond in the π-system of the protonated ring, locally providing aromatic stabilization for the positive charge. The addition of the proton stabilizes all electronic orbitals, but due to the ground state π-electron distribution away from the added nucleus, the HOMO is stabilized less than the LUMO. The main absorption peak upon protonation corresponds to the charge transfer excitation involving the frontier orbitals, and the small band gap explains the observed red shift. Analogue calculations on thiophene within a ZSM-5 zeolite cluster model confirm the same trends upon protonation as observed in the non-interacting compounds. Understanding the electronic structure of these compounds is very relevant to correlate UV-Vis bands with acidic strength and possibly environment in zeolites and to improve their performance in catalytic and energy related applications.
 Please wait while we load your content...
Please wait while we load your content...

