
Size-dependent strain and surface energies of gold nanoclusters†
 b
and
E. C.
Neyts
*a
b
and
E. C.
Neyts
*a
Abstract
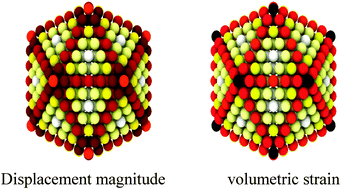
Gold nanocluster properties exhibit unique size-dependence. In this contribution, we employ reactive molecular dynamics simulations to calculate the size- and temperature-dependent surface energies, strain energies and atomic displacements for icosahedral, cuboctahedral, truncated octahedral and decahedral Au-nanoclusters. The calculations demonstrate that the surface energy decreases with increasing cluster size at 0 K but increases with size at higher temperatures. The calculated melting curves as a function of cluster size demonstrate the Gibbs–Thomson effect. Atomic displacements and strain are found to strongly depend on the cluster size and both are found to increase with increasing cluster size. These results are of importance for understanding the size- and temperature-dependent surface processes on gold nanoclusters.
 Please wait while we load your content...
Please wait while we load your content...

