
N-Alkylthienopyrroledione versus benzothiadiazole pulling units in push–pull copolymers used for photovoltaic applications: density functional theory study†
Abstract
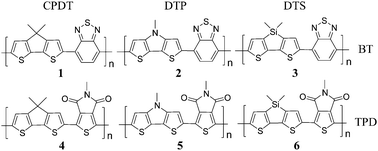
Low-band-gap push–pull copolymers are promising donor materials for bulk heterojunction organic solar cells. One of the best push–pull copolymers are composed of bridged dithiophene pushing units and benzothiadiazole (BT) pulling units, but BT has no proper position to accommodate alkyl side chains introduced to enhance the solubility of the resulting copolymers in organic solvents. On the other hand, N-alkylthienopyrroledione (TPD), which has an alkyl side chain attached to its pyrrole moiety, has been combined with various bridged dithiophene pushing units to give high-solubility donor polymers whose power conversion efficiencies are higher than those of the BT-based polymers especially after a morphology control. However, our well-validated time-dependent density functional theory calculation on the intrinsic (single-chain) electronic structure, which has been proved powerful to estimate the efficiency, gives a contradictory prediction that both polymers would show essentially the same efficiency. Intrigued by this, we subsequently perform density functional theory calculations on their π-stacked-pair models in a number of stacking configurations and conclude that the enhanced performance of the TPD-based polymers is ascribed to their enhanced inter-chain interaction resulting from their enhanced dipole moments in the push–pull direction. Enhanced morphological ordering (π-stacking and π-conjugation) in their solid films, which is not considered in electronic-structure calculations, would reduce the band gap (as proved by the low-energy shoulders in UV/vis absorption spectra), improve the charge transfer (as shown by the calculated transfer integral, transfer rate, and hole mobility), and enhance the power conversion efficiencies (as observed after a morphology control).
 Please wait while we load your content...
Please wait while we load your content...

