
Molecular structure and dynamics of an aqueous sodium chloride solution in nano-pores between portlandite surfaces: a molecular dynamics study
Abstract
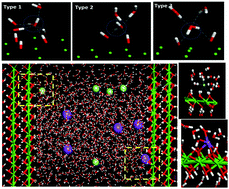
Portlandite plays an important role in the hydration phase of cement-based materials and influences the strength and durability of such materials. This study describes a molecular dynamics study of the structure and dynamics of water and ions confined at ambient temperature in calcium hydroxyl nanopores with widths of 35 Å. Strong layering of water in the vicinity of the (001) surface of portlandite demonstrates special structural features such as large density, good orientation preference, ordered interfacial organization and low diffusion rate. Due to the fixed vibration and rotation of the hydroxyl groups at the interface, water molecules within the first adsorbed layer adopt both H-downward and H-upward orientations by donating H-bonds and accepting H-bonds from the OH groups in the solid surface. Regarding the interaction of the ions and portlandite, Na+ ions, deeply rooted in spaces in the surface hydroxyl groups, are significantly slowed and remain near the surface for long periods of time. On the other hand, due to the weak H-bonds formed by chloride ions and hydroxyl groups, adsorbed chloride ions near the surface cannot remain for longer times. In addition, when water and ions are confined in the nano-pores, the residence time for the ion–water and ion–ion clusters is lengthened so that the ion adsorption capability of the porlandite surface is enhanced due to the stable Na–Cl connections in the electrolyte solution.
 Please wait while we load your content...
Please wait while we load your content...

