
NO adsorption and diffusion on hydroxylated rutile TiO2(110)†
Abstract
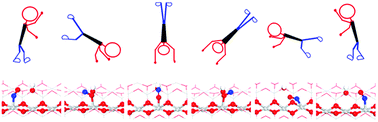
We report a computational study of NO adsorption and diffusion on the hydroxylated rutile TiO2(110) surface performed with density functional theory (DFT) calculations corrected by on-site Coulomb corrections and long-range dispersion interactions. NO prefers to adsorb with its N-end down at surface Ti5c sites. The excess electron that is located at a subsurface site for the hydroxylated surface localizes in the 2π* orbital of the adsorbed NO. A novel ‘roll-over’ diffusion scheme is proposed that involves three neighboring Ti5c atoms and one surface hydroxyl, with an O-end down NO at the middle Ti5c as the intermediate state. During the migration, NO can also form bridging species between two Ti5c atoms. The calculated scanning tunneling microscopy (STM) features with the “bright-dark-bright” configuration corresponding to diffusing NO at different positions are consistent with the experimental STM results.
 Please wait while we load your content...
Please wait while we load your content...

