
A comparison of ab initio quantum-mechanical and experimental D0 binding energies of eleven H-bonded and eleven dispersion-bound complexes
Abstract
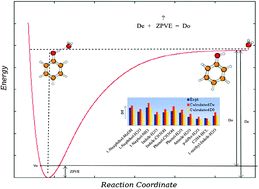
Dissociation energies (D0) of 11 H-bonded and 11 dispersion-bound complexes were calculated as the sum of interaction energies and the change of zero-point vibrational energies (ΔZPVE). The structures of H-bonded complexes were optimized at the RI-MP2/cc-pVTZ level, at which deformation and harmonic ΔZPVE energies were also calculated. The structures of dispersion-bound complexes were optimized at the DFT-D3 level, and harmonic ΔZPVE energies were determined at the same level as well. For comparison, CCSD(T)/CBS D0 energies were also evaluated for both types of complexes. The CCSD(T)/CBS interaction energy was constructed as the sum of MP2/CBS interaction energy, extrapolated from aug-cc-pVTZ and aug-cc-pVQZ basis sets, and ΔCCSD(T) correction, determined with the aug-cc-pVDZ basis set. The ΔZPVE energies were determined for all complexes at the harmonic level and for selected complexes, these energies were also calculated using second-order vibration perturbation (VPT2) theory. For H-bonded complexes, the harmonic CCSD(T)/CBS D0 energies were in better agreement with the experimental values (with a mean relative error (MRE) of 6.2%) than the RI-MP2/cc-pVTZ D0 (a MRE of 12.3%). The same trend was found for dispersion-bound complexes (6.2% (MRE) at CCSD(T)/CBS and 7.7% (MRE) at the DFT-D3 level). When the anharmonic ΔZPVE term was included instead of harmonic one, the agreement between theoretical and experimental D0 deteriorated for H-bonded as well as dispersion-bound complexes. Finally, the applicability of “diagonal approximation” for determining the anharmonic ΔZPVE was shown. For the phenol⋯H2O complex, the ΔZPVE energy calculated at the VPT2 level and on the basis of “diagonal approximation” differed by less than 0.1 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

