
Position and orientational preferences of drug-like compounds in lipid membranes: a computational and NMR approach†
Abstract
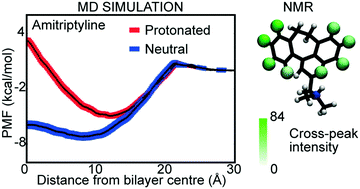
Permeation of drugs across lipid bilayers is a key factor in dictating how effective they will be. In vivo, the issue is compounded by the presence of drug–exporter proteins such as P-glycoprotein. However, despite intense effort, exactly what controls permeation and susceptibility to export is still poorly understood. In this work we examine two well-studied drugs for which interaction with P-glycoprotein has been studied before: amitriptyline, a known substrate and clozapine, which is not a substrate. Extensive MD simulations, including potential of mean force (PMF) profiles of the compounds in all possible protonation states, reveal that the preferred location of the compounds in different bilayers in different protonation states is remarkably similar. For both molecules in charged states, there is a substantial barrier to crossing the bilayer. Clozapine however, shows an energetic barrier to movement across the bilayer even in a protonation state that results in an uncharged molecule. For amitriptyline there is only a very small barrier of approximately 1.3 kcal mol−1. Further analysis revealed that the conformational and orientational behavior of the two compounds was also similar, with the sidechain interacting with the lipid headgroups. This effect was much stronger if the sidechain was charged (protonated). These interactions with lipid bilayers were confirmed by NMR ROESY experiments. The results are discussed in terms of their potential interactions with export proteins like P-glycoprotein.
 Please wait while we load your content...
Please wait while we load your content...

