
An ultrafast spectroscopic and quantum mechanical investigation of multiple emissions in push–pull pyridinium derivatives bearing different electron donors
Abstract
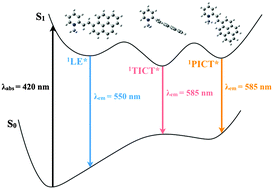
A joint experimental and theoretical approach, involving state-of-the-art femtosecond fluorescence up-conversion measurements and quantum mechanical computations including vibronic effects, was employed to get a deep insight into the excited state dynamics of two cationic dipolar chromophores (Donor–π–Acceptor+) where the electron deficient portion is a N-methyl pyridinium and the electron donor a trimethoxyphenyl or a pyrene, respectively. The ultrafast spectroscopic investigation, and the time resolved area normalised emission spectra in particular, revealed a peculiar multiple emissive behaviour and allowed the distinct emitting states to be remarkably distinguished from solvation dynamics, occurring in water in a similar timescale. The two and three emissions experimentally detected for the trimethoxyphenyl and pyrene derivatives, respectively, were associated with specific local emissive minima in the potential energy surface of S1 on the ground of quantum–mechanical calculations. A low polar and planar Locally Excited (LE) state together with a highly polar and Twisted Intramolecular Charge Transfer (TICT) state is identified to be responsible for the dual emission of the trimethoxyphenyl compound. Interestingly, the more complex photobehaviour of the pyrenyl derivative was explained considering the contribution to the fluorescence coming not only from the LE and TICT states but also from a nearly Planar Intramolecular Charge Transfer (PICT) state, with both the TICT and the PICT generated from LE by progressive torsion around the quasi-single bond between the methylpyridinium and the ethene bridge. These findings point to an interconversion between rotamers for the pyrene compound taking place in its excited state against the Non-equilibrated Excited Rotamers (NEER) principle.
 Please wait while we load your content...
Please wait while we load your content...

