
DFT studies of the bonding mechanism of 8-hydroxyquinoline and derivatives on the (111) aluminum surface
Abstract
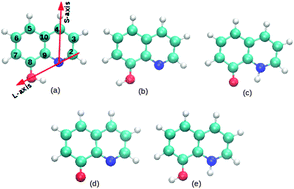
The 8-hydroxyquinoline (8-HQ) molecule is an efficient corrosion inhibitor for aluminum and is also used in organic electronic devices. In this paper, the adsorption modes of 8-HQ and its derivatives (tautomer, dehydrogenated and hydrogenated species) on the Al(111) surface are characterized using dispersion corrected density functional theory calculations. The 8-HQ molecule is physisorbed and is chemisorbed on the aluminum surface with similar adsorption energy (−0.86 eV to −1.11 eV) and these adsorption modes are stabilized by vdW interactions. The binding of the dehydrogenated species is the strongest one (adsorption energy of −3.27 eV to −3.45 eV), followed by the tautomer molecule (−2.16 eV to −2.39 eV) and the hydrogenated molecule (−1.71 eV) that bind weaker. In all the chemisorbed configurations there is a strong electronic transfer from the Al substrate to the adsorbate (0.72 e to 2.16 e). The adsorbate is strongly distorted and its deformation energy is high (0.55 eV to 2.77 eV). The analysis of the projected density of states onto the orbitals of the molecule and the electronic density variation upon adsorption (Δρ) between the molecule and the surface account for covalent bonding.
 Please wait while we load your content...
Please wait while we load your content...

