
Corrole–ferrocene and corrole–anthraquinone dyads: synthesis, spectroscopy and photochemistry†
Abstract
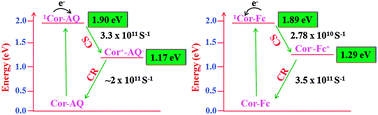
Two different donor–acceptor systems based on corrole–ferrocene and corrole–anthraquinone having the ‘Olefin Bridge’ at the β-pyrrole position have been designed and synthesized. Both the dyads corrole–ferrocene (Cor–Fc) and corrole–anthraquinone (Cor–AQ) are characterized by elemental analysis, ESI-MS, 1H NMR, UV-Visible, fluorescence spectroscopies (steady-state, femtosecond time-resolved), femtosecond transient absorption spectroscopy (fs-TA) and electrochemical methods. 1H-NMR shows that two doublets at 6.50 and 7.25(δ) ppm belong to vinylic protons, which are characteristic of the formation of dyads. UV-Visible absorption spectra showed that dyads are merely superpositions of their respective constituent monomers and dominated by corrole S1 ← S0 (Q-band) and S2 ← S0 (Soret band) transitions with a systematic red-shift of both Soret and Q-bands along with the broadening of the bands. A prominent splitting of the Soret band for both the dyads is observed due to bulky substitutions at the peripheral position, which deviate from the planarity of the corrole macrocycle. Both the dyads exhibit significant fluorescence emission quenching (95–97%) of corrole emission compared to the free-base corrole monomer. Emission quenching is attributed to the excited-state intramolecular photoinduced electron transfer (PET) from corrole to anthraquinone in the Cor–AQ dyad, whereas in the Cor–Fc dyad it is reversed. The electron-transfer rates (kET) for Cor–AQ and Cor–Fc were found to be 3.33 × 1011 and 2.78 × 1010 s−1, respectively. Despite their very different driving forces, charge separation (CS) and charge recombination (CR) are found to be in identical timescales.
 Please wait while we load your content...
Please wait while we load your content...

