
Coordination properties of a metal chelator clioquinol to Zn2+ studied by static DFT and ab initio molecular dynamics†
Abstract
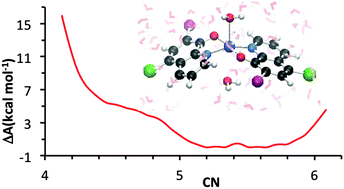
Several lines of evidence supporting the role of metal ions in amyloid aggregation, one of the hallmarks of Alzheimer's disease (AD), have turned metal ion chelation into a promising therapeutic treatment. The design of efficient chelating ligands requires proper knowledge of the electronic and molecular structure of the complexes formed, including their hydration properties. Among various potential chelators, clioquinol (5-chloro-7-iodo-8-hydroxyquinoline, CQH) has been evaluated with relative success in in vitro experiments and even in phase 2 clinical trials. Clioquinol interacts with Zn(II) to lead to a binary metal/ligand 1 : 2 stoichiometric complex in which the phenolic group of CQH is deprotonated, resulting in Zn(CQ)2 neutral complexes, to which additional water molecules may coordinate. In the present work, the coordinative properties of clioquinol in aqueous solution have been analyzed by means of static, minimal cluster based DFT calculations and explicit solvent ab initio molecular dynamics simulations. Results from static calculations accounting for solvent effects by means of polarized continuum models suggest that the preferred metal coordination environment is tetrahedral Zn(CQ)2, whereas ab initio molecular dynamics simulations point to quasi degenerate penta Zn(CQ)2(H2O) and hexa Zn(CQ)2(H2O)2 coordinated complexes. The possible reasons for these discrepant results are discussed.
 Please wait while we load your content...
Please wait while we load your content...

