
A first-principles study on the magnetic properties of nonmetal atom doped phosphorene monolayers†
Abstract
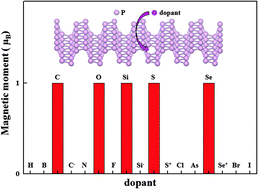
In order to induce magnetism in two-dimensional semiconductors for their applications in spintronic devices and novel chemical and electronic properties of semiconducting phosphorene, the geometrical structure, electronic and magnetic properties of doped phosphorene monolayers with a series of nonmetal atoms, including H, F, Cl, Br, I, B, C, Si, N, As, O, S and Se, were systematically investigated using first-principles calculations. The results show that although the substitutional doping of H, F, Cl, Br, I, B, N, O, S or Se results in large structural deformation at the doping sites of phosphorene monolayers, all neutral nonmetal atom doped systems are stable. The calculated formation energies reveal that the substitutional doping of numerous nonmetal atoms in phosphorene monolayer are possible under appropriate experimental conditions, and the charged dopants C−, Si−, S+ and Se+ are stable. Moreover, the substitutional doping of H, F, Cl, Br, I, B, N, As, C−, Si−, S+ or Se+ cannot induce magnetism in phosphorene monolayer due to the saturation or pairing of valence electrons of dopant and its neighboring P atoms, whereas ground states of neutral C, Si, O, S or Se doped systems are magnetic due to the appearance of an unpaired valence electron of C and Si or the formation of a nonbonding 3p electron of a neighboring P atom around O, S and Se. Furthermore, the magnetic coupling between the moments induced by two Si, O, S or Se are long-range anti-ferromagnetic and the coupling can be attributed to the hybridization interaction involving polarized electrons, whereas the coupling between the moments induced by two C is weak.
 Please wait while we load your content...
Please wait while we load your content...

