
Borazine: spin blocker or not?†
Abstract
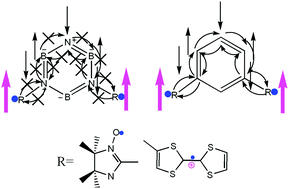
The spin blocker capacity of borazine is investigated. Specifically, meta-B–B, meta-N–N and para-B–N connected borazines are used as spin-blocker couplers comprised of a pair of radicals: two iminonitroxides (IN); IN and tetrathiafulvalene radical cations (TTF); or two TTFs. Density functional theory (DFT) is used to elucidate the spin blocker capacity of the linkage-specific (meta or para) borazine-coupler and elaborate the role of the lowest unoccupied molecular orbital (LUMO) in magnetic–exchange. Furthermore, a qualitative relation between different magnetic aromaticity indices is made using both nuclear-independent chemical shift (NICS) and the harmonic oscillator model of aromaticity (HOMA). The NICS values are calculated at the centre of the borazine spacer fragment of these diradical species and then also at 0.5 Å increments of the virtual probe from this centre position up to an orthogonal distance of 2.0 Å from the centre. The HOMA values are calculated for the borazine ring fragment in these diradicals. Based on the HOMA and NICS values, it is evident that the borazine exhibits less aromatic character than benzene itself – due to the polar nature of B–N π-bonding. The LUMO mediated spin-exchange between the two consecutive singly occupied molecular orbitals (SOMOs) is explicitly discussed and confirmed to play a pivotal role. The parity of the coupler pathways, i.e. even or odd number of bonds along a selected pathway, between radical moieties is an important factor in predicting the nature and extent of magnetic exchange for these diradicals. Surprisingly, borazine does not always act as a spin-coupling blocker – rather in some cases the coupling is enhanced as compared to a homoatomic (carbon-based) benzene coupler.
 Please wait while we load your content...
Please wait while we load your content...

