
Modeling ultrafast exciton deactivation in oligothiophenes via nonadiabatic dynamics†
Abstract
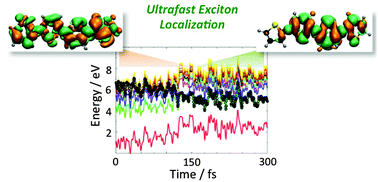
Ultrafast excited-state processes play a key role in organic electronics and photovoltaics, governing the way of how excitons can relax and separate. Through the use of nonadiabatic excited-state dynamics, relaxation processes were investigated at the sub-picosecond timescale in thiophene and oligothiophenes (nT, n = 2, 3, 4), prototype oligomers for efficient π-electron conjugated polymers adopted in photovoltaics. For thiophene, TDDFT and TDA nonadiabatic excited-state dynamics revealed ultrafast nonradiative relaxation processes through ring opening and ring puckering, bringing the system to an S1/S0 conical intersection seam. The computed relaxation time is 110 fs, matching well the experimental one (∼105 fs). In oligothiophenes (n = 2–4), high-energy (hot) excitations were considered. Exciton relaxation through the manifold of excited states to the lowest excited state is predicted to occur within ∼150–200 fs, involving bond stretching, ring puckering, and torsional oscillations. For the longer oligomer (4T), the ultrafast relaxation process leads to exciton localization over three thiophene rings in 150 fs. These data agree with the self-localization mechanism (∼100–200 fs) observed for poly(3-hexylthiophene) (P3HT) and shed light on the complex exciton relaxation dynamics occurring in π-conjugated oligomers of potential interest for optoelectronic applications.
 Please wait while we load your content...
Please wait while we load your content...

