
Analysis of the conformational profiles of fenamates shows route towards novel, higher accuracy, force-fields for pharmaceuticals†
Abstract
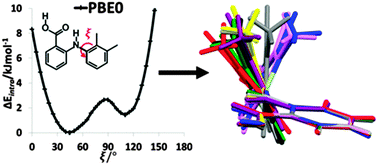
In traditional molecular mechanics force fields, intramolecular non-bonded interactions are modelled as intermolecular interactions, and the form of the torsion potential is based on the conformational profiles of small organic molecules. We investigate how a separate model for the intramolecular forces in pharmaceuticals could be more realistic by analysing the low barrier to rotation of the phenyl ring in the fenamates (substituted N-phenyl-aminobenzoic acids), that results in a wide range of observed angles in the numerous fenamate crystal structures. Although the conformational energy changes by significantly less than 10 kJ mol−1 for a complete rotation of the phenyl ring for fenamic acid, the barrier is only small because of small correlated changes in the other bond and torsion angles. The maxima for conformations where the two aromatic rings approach coplanarity arise from steric repulsion, but the maxima when the two rings are approximately perpendicular arise from a combination of an electronic effect and intramolecular dispersion. Representing the ab initio conformational energy profiles as a cosine series alone is ineffective; however, combining a cos 2ξ term to represent the electronic barrier with an intramolecular atom–atom exp-6 term for all atom pairs separated by three or more bonds (1–4 interactions) provides a very effective representation. Thus we propose a new, physically motivated, generic analytical model of conformational energy, which could be combined with an intermolecular model to form more accurate force-fields for modelling the condensed phases of pharmaceutical-like organic molecules.
 Please wait while we load your content...
Please wait while we load your content...

