
Subsystem-DFT potential-energy curves for weakly interacting systems†
Abstract
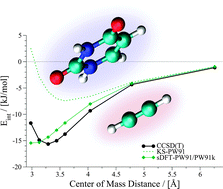
Kohn–Sham density-functional theory (DFT) within the local-density approximation (LDA) or the generalized-gradient approximation (GGA) is known to fail for the correct description of London dispersion interactions. Often, not even bound potential-energy surfaces are obtained for van der Waals complexes, unless special correction schemes are employed. In contrast to that, there has been some evidence for the fact that subsystem-based density functional theory produces interaction energies for weakly bound systems which are superior to Kohn–Sham DFT results without dispersion corrections. This is usually attributed to an error cancellation between the approximate exchange–correlation and non-additive kinetic-energy functionals employed in subsystem DFT. Here, we investigate the accuracy of subsystem DFT for weakly interacting systems in detail, paying special attention to the shape of the potential-energy surfaces (PESs). Our test sets include the extensive S22x5 and S66x8 data sets. Our results indicate that subsystem DFT PESs strongly vary depending on the functional. LDA results are usually quite good, but behave differently from their KS counterparts. GGA results from the popular Perdew–Wang (PW91) set of functionals produce PESs that are often, but not in general overbinding. Results from Becke–Perdew (BP86) GGAs, by contrast, show the typical problems known from the corresponding KS results. We provide some preliminary results for empirical corrections for both PW91 and BP86 in subsystem DFT.
- This article is part of the themed collection: Theoretical chemistry developments: from electronic structure to simulations
 Please wait while we load your content...
Please wait while we load your content...

