
Computational study on the interaction between CCR5 and HIV-1 entry inhibitor maraviroc: insight from accelerated molecular dynamics simulation and free energy calculation†
Abstract
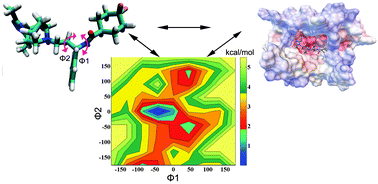
C–C chemokine receptor type 5 (CCR5) is the co-receptor of human immunodeficiency virus type 1 (HIV-1) and plays an important role in HIV-1 virus infection. Maraviroc has been proved to be effective for anti-HIV-1 by targeting CCR5. Understanding the detailed interaction mechanism between CCR5 and Maraviroc will be of great help to the rational design of a more potential inverse agonist to block HIV-1 infection. Here, we performed molecular dynamics (MD) simulation and accelerated MD simulation (aMD) to study the interaction mechanism between CCR5 and Maraviroc based on a recently reported crystal structure. The results of MD simulation demonstrate that Maraviroc can form stable hydrogen bonds with residues Tyr371.39, Tyr2516.51 and Glu2837.39. The results of aMD simulation indicate that the carboxamide moiety is more flexible than the tropane group of Maraviroc in the pocket of CCR5. The electrostatic potential analysis proves that Maraviroc can escape from the pocket of CCR5 along the negative electrostatic potential pathway during the dissociation process. The free energy calculation illustrates that there exist three binding pockets during the dissociation process of Maraviroc. Our results will be useful for understanding the interaction mechanism between CCR5 and Maraviroc as well as for the rational design of a more potent inverse agonist.
 Please wait while we load your content...
Please wait while we load your content...

