
A density function theory study on the NO reduction on nitrogen doped graphene†
Abstract
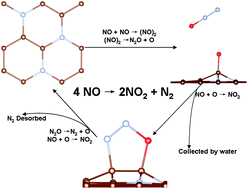
The mechanisms for the catalytic reduction of NO on the metal-free nitrogen doped graphene (NG) support are investigated using the density function theory (DFT) calculations both with and without the van der Waals (vdW) correction. The results indicate that the dimer mechanism is more facile than the direct decomposition mechanism. In the dimer mechanism, a three-step reaction is identified: (i) the coupling of two NO molecules into a (NO)2 dimer, followed by (ii) the dissociation of the (NO)2 dimer into N2O + Oad, then (iii) the O adatom is taken away easily by the subsequent NO. Once the NO2 is desorbed, the remaining N2O can be reduced readily by NO on NG. The reaction processes are also confirmed from the first principles molecular dynamics simulations. The results suggest that the NG is an efficient metal-free catalyst for catalytic reduction of NO.
 Please wait while we load your content...
Please wait while we load your content...

