
The structural origin of the unusual compression behaviors in nanostructured TiO2: insights from first-principles calculations
Abstract
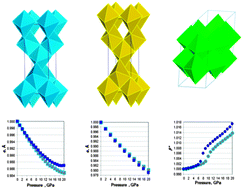
First-principles calculations of anatase structured TiO2 and ZrO2 as well as of TiO2–B were carried up to 20 GPa in order to develop an understanding of the unusual compression and pressure-dependent phase transitions reported for nanocrystalline (nc) pure and Zr-doped anatase and nc TiO2–B. The computations, carried out using two global hybrid density functional-Hartree–Fock formulations and all-electron basis sets, reveal sharp lattice hardening along the crystallographic a direction and concurrent lattice softening along c for anatase TiO2 at 10–12 GPa, and smooth anisotropic compression for ZrO2 anatase. Significant structural changes beginning at ∼10 GPa are also predicted for TiO2–B, most dramatically shown by the pressure-dependent change in the monoclinic angle β. These structural changes, resulting from intrinsic crystal structure destabilization under extended pressure metastability, have been suggested as being responsible for the unusual mechanical behaviors reported for pure and Zr-doped nanocrystalline and microcrystalline anatase TiO2 and nc TiO2–B.
 Please wait while we load your content...
Please wait while we load your content...

