
Superbasicity of silylene derivatives achieved via non-covalent intramolecular cation⋯π interactions and exploited as molecular containers for CO2†
Abstract
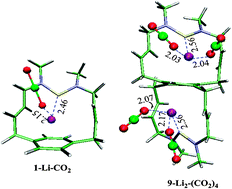
We have reported for the first time designed silylene superbases involving intramolecular H+⋯π interaction using density functional theory (DFT) calculations. The non-covalent interactions augment the proton affinity values by ∼13 kcal mol−1 in the designed superbase (1) compared to the acyclic silylene, [:Si(NMe2)2]. These divalent Si(II) compounds can act as powerful neutral organic superbases in gas and solvent phases. The DFT calculations performed at the B3LYP/6-311+G**//B3LYP/6-31+G* level of theory showed that the gas phase proton affinity of the paracyclophane based silylene superbase (8) reaches up to ∼271.0 kcal mol−1, which is the highest in paracyclophane Si(II) compounds. In tetrahydrofuran solvent medium, the calculated proton affinity of 8 was found to be 301.4 kcal mol−1. The paracyclophane-based silylene systems are used for binding with small alkali metal ions. The calculated results showed that these systems selectively bind to lithium ions over sodium ions due to the small size of lithium ions which is well fitted in the space between the silicon atom and the phenyl ring. Furthermore, we have used the lithiated silylene 1 and 9 (which exhibits bis-protonation) for gas storage (CO and CO2). The calculated results showed both the lithiated silylene 1 and 9 bind preferentially to CO2 than CO. The calculated gravimetric density of CO2 is found to be 26.97 wt% for 9-Li2–(CO2)4. The energy decomposition analysis (EDA) has been performed to investigate the role of various contributing factors to the total binding strength of the CO2 or CO molecules with lithiated silylene superbases. EDA reveals that the electrostatic energy and polarization energy are the major driving force for higher total interaction energy of the lithiated-silylene–CO2 complex than the lithiated-silylene–CO complex. The lithiated silylene systems showed a higher binding energy with CO2 than the previously reported imidazopyridamine at the same level of theory. These results suggest that the lithiated silylene systems can be used as a more efficient CO2 storage material than the aforementioned system. The calculated desorption energies per CO2 and CO (ΔEDE) also indicate the recyclable property of the materials.
 Please wait while we load your content...
Please wait while we load your content...

