
Ab initio quantum mechanical simulations confirm the formation of all postulated species in ionic dissociation
Abstract
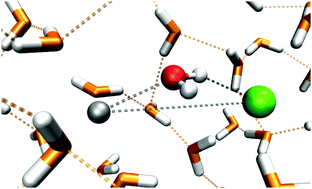
A single sodium chloride molecule in aqueous solution was simulated by the ab initio quantum mechanical charge field-molecular dynamics (QMCF-MD) approach. During a series of simulations the solvated molecule (CIP), dissociated solvated ions and – most noticeably – a solvent separated ion pair (SSIP) were observed and the structural and dynamical characteristics of these systems were investigated. In addition to a detailed structural analysis of the observed species, vibrational spectra and charge distributions were calculated to elucidate the mechanism of the NaCl dissociation.
 Please wait while we load your content...
Please wait while we load your content...

