
Ab initio non-adiabatic molecular dynamics
Abstract
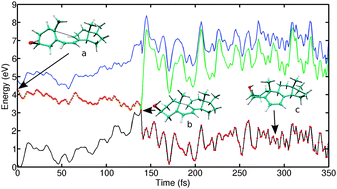
Adiabatic nuclear potential energy surfaces (PESs) defined via the Born–Oppenheimer (BO) approximation are a fundamental concept underlying chemical reactivity theory. For a wide range of excited-state phenomena such as radiationless decay, energy and charge transfer, and photochemical reactions, the BO approximation breaks down due to strong couplings between two or more BO PESs. Non-adiabatic molecular dynamics (NAMD) is the method of choice to model these processes. We review new developments in quantum–classical dynamics, analytical derivative methods, and time-dependent density functional theory (TDDFT) which have lead to a dramatic expansion of the scope of ab initio NAMD simulations for molecular systems in recent years. We focus on atom-centered Gaussian basis sets allowing highly efficient simulations for molecules and clusters, especially in conjunction with hybrid density functionals. Using analytical derivative techniques, forces and derivative couplings can be obtained with machine precision in a given basis set, which is crucial for accurate and stable dynamics. We illustrate the performance of surface-hopping TDDFT for photochemical reactions of the lowest singlet excited states of cyclohexadiene, several vitamin D derivatives, and a bicyclic cyclobutene. With few exceptions, the calculated quantum yields and excited state lifetimes agree qualitatively with experiment. For systems with ∼50 atoms, the present TURBOMOLE implementation allows NAMD simulations with 0.2–0.4 ns total simulation time using hybrid density functionals and polarized double zeta valence basis sets on medium-size compute clusters. We close by discussing open problems and future directions.
 Please wait while we load your content...
Please wait while we load your content...

