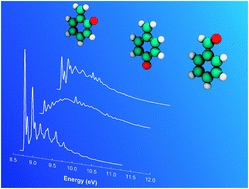
The resonance stabilized benzyl radical is an important intermediate in the combustion of aromatic hydrocarbons and in polycyclic aromatic hydrocarbon (PAH) formation in flames. Despite being a free radical, benzyl is relatively stable in thermal, oxidizing environments, and is predominantly removed through bimolecular reactions with open-shell species other than O2. In this study the reaction of benzyl with ground-state atomic oxygen, O(3P), is examined using quantum chemistry and statistical reaction rate theory. C7H7O energy surfaces are generated at the G3SX level, and include several novel pathways. Transition state theory is used to describe elementary reaction kinetics, with canonical variational transition state theory applied for barrierless O atom association with benzyl. Apparent rate constants and branching ratios to different product sets are obtained as a function of temperature and pressure from solving the time-dependent master equation, with RRKM theory for microcanonical k(E). These simulations indicate that the benzyl + O reaction predominantly forms the phenyl radical (C6H5) plus formaldehyde (HCHO), with lesser quantities of the C7H6O products benzaldehyde, ortho-quinone methide, and para-quinone methide (+H), along with minor amounts of the formyl radical (HCO) + benzene. Addition of O(3P) to the methylene site in benzyl produces a highly vibrationally excited C7H7O* adduct, the benzoxyl radical, which can β-scission to benzaldehyde + H and phenyl + HCHO. In order to account for the experimental observation of benzene as the major reaction product, a roaming radical mechanism is proposed that converts the nascent products phenyl and HCHO to benzene + HCO. Oxygen atom addition at the ortho and para ring sites in benzyl, which has not been previously considered, is shown to lead to the quinone methides + H; these species are less-stable isomers of benzaldehyde that are proposed as important combustion intermediates, but are yet to be identified experimentally. Franck–Condon simulations of the benzaldehyde, o-quinone methide, and p-quinone methide photoelectron spectra suggest that these C7H6O isomers could be distinguished using tunable VUV photoionization mass spectrometry.

 Please wait while we load your content...
Please wait while we load your content...

