
Quantum dynamics study of the Cl + CH4 → HCl + CH3 reaction: reactive resonance, vibrational excitation reactivity, and rate constants
Abstract
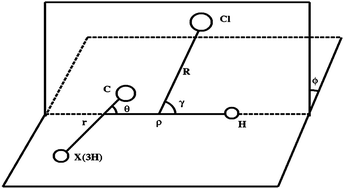
A quantum reactive dynamics, six-degrees-of-freedom, time-dependent wavepacket propagation method is applied to study the Cl + CH4 → HCl + CH3 reaction on the newly published potential energy surface by Czakó and Bowman [Science, 2011, 334, 343; J. Chem. Phys., 2012, 136, 044307]. We confirm not only the experimental speculation of the reactive resonance by observing a prominent resonance peak on the ground state reaction probability, but also the experimental and quasi-classical trajectory finding that at lower total scattering energy the translational energy drives the reactivity more than the vibrational energy for this late barrier reaction. The vibrational motions of CH4 enhance the reactivity, and the C–H stretching motion has the biggest impact on the reactivity. The vibrational energy overall plays a more efficient role in the reactivity than the translational energy except at the lower scattering energy. The energy-shift approximation is employed to obtain an approximate full-dimensional cumulative reaction probability based on the six dimensional calculation. The calculated thermal rate coefficients agree very well with experimental measurements after using experimental vibrational frequencies and zero point energy to correct the reactant vibrational partition function and to convert the energy for the full dimensional cumulative reaction probability.
 Please wait while we load your content...
Please wait while we load your content...

