
Contrasting preferences of N and P substituted heteroaromatics towards metal binding: probing the regioselectivity of Li+ and Mg2+ binding to (CH)6−m−nNmPn†
Abstract
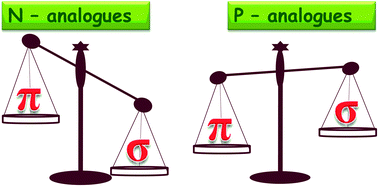
High level ab initio and hybrid DFT methods have been employed to investigate the interactions of metal ions (Li+ and Mg2+) with N and P substituted six membered heteroaromatics (CH)6−m−nNmPn. The binding energy (BE) of metal ions with the N and P substituted heteroaromatics has been computed at the CCSD(T)/cc-pVTZ//MP2/cc-pVTZ level with counterpoise correction. In the present study we systematically examined the preferential modes of binding of metal ions to the heteroaromatics. N-Substituted heteroaromatics show a strong preference for cation–σ mode of binding whereas the P-substituted heteroaromatics prefer cation–π mode of binding with the metal ions. Energy decomposition analysis (EDA) using the DFT-SAPT scheme has been carried out to analyse the contribution of various energy components to the BE. The results illustrate that for the cation–π complexes, the contribution of the induction term is more whereas in the case of cation–σ there is a competition between induction and electrostatic terms in the interaction energy.
 Please wait while we load your content...
Please wait while we load your content...

