
Liquid water simulations with the density fragment interaction approach†
Abstract
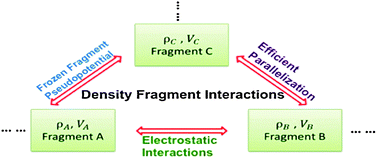
We reformulate the density fragment interaction (DFI) approach [Fujimoto and Yang, J. Chem. Phys., 2008, 129, 054102.] to achieve linear-scaling quantum mechanical calculations for large molecular systems. Two key approximations are developed to improve the efficiency of the DFI approach and thus enable the calculations for large molecules: the electrostatic interactions between fragments are computed efficiently by means of polarizable electrostatic-potential-fitted atomic charges; and frozen fragment pseudopotentials, similar to the effective fragment potentials that can be fitted from interactions between small molecules, are employed to take into account the Pauli repulsion effect among fragments. Our reformulated and parallelized DFI method demonstrates excellent parallel performance based on the benchmarks for the system of 256
- This article is part of the themed collection: Fragment and localized orbital methods in electronic structure theory
 Please wait while we load your content...
Please wait while we load your content...

