
Comparison of the performance of dispersion-corrected density functional theory for weak hydrogen bonds†
Abstract
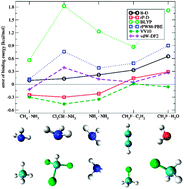
Potential energy curves for five complexes with weak to medium strong hydrogen bonds have been computed with dispersion corrected DFT methods. The electronic density based vdW-DF2 and VV10 van der Waals density functionals have been tested, as well as an atom pair-wise correction method (DFT-D3). The short-range exchange–correlation components BLYP and rPW86-PBE together with the extended aug-cc-pVQZ basis sets have been employed. Reference data have been computed at the estimated CCSD(T)/CBS(aQ-a5) level of theory. The investigated systems are CH4·NH3, Cl3CH·NH3, NH3·NH3, CH3F·C2H2 and CH3F·H2O with binding energies ranging from −0.7 kcal mol−1 to −5.5 kcal mol−1. We find that all dispersion corrected methods perform reasonably well for these hydrogen bonds, but also observe distinct differences. The BLYP-D3 method provides the best results for three out of five systems. For the fluorinated complexes, the VV10 method gives remarkably good results. The vdW-DF2 method yields good interaction energies similar to the other methods (mean average deviation of 0.2–0.3 kcal mol−1), but fails to provide accurate equilibrium separations. Based on these results and previous experience with the computation of non-covalent interactions, for large-scale applications we can recommend DFT-D3 based structure optimizations with subsequent checking of interaction energies by single-point VV10 computations. Comparison of the DFT-D3 and VV10 results leads to the conclusion that the short-range exchange–correlation functional and not the dispersion correction mainly determines the achievable accuracy.
- This article is part of the themed collection: Weak hydrogen bonds – strong effects?
 Please wait while we load your content...
Please wait while we load your content...

