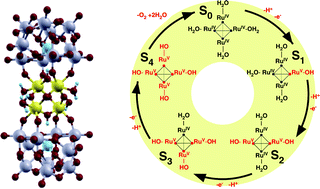
We present a computational study addressing the catalytic cycle of a recently-synthesized all-inorganic homogeneous catalyst capable to promote water oxidation with low overpotential and high turnover frequency [Sartorel et al., J. Am. Chem. Soc., 2008, 130, 5006; Geletii et al., Angew. Chem., Int. Ed., 2008, 47, 3896]. This catalyst consists of a tetraruthenium-oxo core [Ru4O4(OH)2·(H2O)4]6+capped by two polyoxometalate [SiW10O36]8− units. The reaction mechanism underpinning its efficiency is currently under debate. We study a reaction cycle involving four consecutive proton-coupled electron transfer (PCET) processes that successively oxidize the four RuIV–H2O units of the initial state (S0) to the four RuV-OH centers of the activated intermediate (S4). The energetics of these electrochemical processes as well as the structural and electronic properties of the reaction intermediates are studied with ab initio Density Functional Theory (DFT) calculations. After characterizing these reaction intermediates in the gas phase, we show that the solvated tetraruthenate core undergoes a solvent-induced structural distortion that brings the predicted molecular geometry to excellent agreement with the experimental X-ray diffraction data. The calculated electronic properties of the catalyst are instead weakly dependent on the presence of the solvent. The frontier orbitals of the initial state as well as the electronic states involved in the PCET steps are shown to be localized on the tetraruthenium-oxo core. The reaction thermodynamics predicted for the intermediate reaction steps is in good agreement with the available cyclic voltammetry measurements up to S3, but the calculated free energy difference between the initial and the activated state (S0/S4) turns out to be significantly lower than the thermodynamic limit for water oxidation. Since the oxidizing power of the S0/S4 couple is not sufficient to split water, we suggest that promoting this reaction would require cycling between higher oxidation states.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


