
Molecular simulation of conformational transitions in biomolecules using a combination of structure-based potential and empirical valence bond theory†
Abstract
The functions of biological
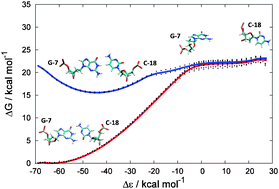
- This article is part of the themed collection: Nucleic acid simulations
 Please wait while we load your content...
Please wait while we load your content...

